Heck, on a rare day you may actually have 3 or 4 patients with alcohol intoxication!
 |
| This is a thing that happens. Source |
Last week for example, the theme for Yuko & I was narrow-complex tachycardias. As themes go, this was a good one - quick procedures, no untoward events, and we made the patients felt better!
Mrs Black
An older lady, Mrs Black had felt palpitations start about an hour before she arrived in the ED. Although she had no chest pain or pressure, she admitted to a little shortness of breath, as well as mild nausea. She took a variety of medications for hypertension, but no calcium-channel blockers or beta-blockers. Her vitals were normal.
Her first ECG:
 |
| Mrs. Black - before |
After 6 mg of adenosine, we had:
 |
| Mrs. Black - after |
Mr White
Our next PSVT came in only 2 hours later.
Also elderly, Mr White had been having some mild racing of his pulse over the past week, but the symptoms acutely worsened about 3 hours prior to coming to the ED. He had used a home-BP machine and noted that his systolic pressure had only been 70 at one point. The ED tech, of note, mentioned that he had looked fairly ill at triage - pale and sweaty.
His first ECG:
 |
| Mr White - before |
And again, after 6 mg of adenosine (I'm not very original):
 |
| Mr White - after |
So what's going on?
Ok, they're both narrow-complex tachycardias, very regular, with rates well under 150. So atrial fibrillation and flutter are pretty unlikely, as is a sinus tachycardia.
So that leaves the somewhat broad category of PSVT. Now, we usually just leave it at that in the ED, because, well, it doesn't usually matter to us whether the rhythm is caused by intranodal reentry (as in AV nodal reentrant tachycardia, or AVNRT) or extranodal reentry (as in AV reentrant tachycardia, or AVRT). This family of arrhythmias is almost always safe, even if the ECG shows ST depression, or we find a small troponin bump after conversion.
But the differentiation between different types of PSVTs is very important to the cardiologist, as there are implications for therapy. For example, AVRT is almost always primarily treated with an ablation. AVNRT, however, often is first treated a trial of medications (such as calcium-channel blockers, beta-blockers, or even digoxin), although ablation remains an option.
So when one of the new cardiologists, Ram Gordon, from Cardiac Specialists, pointed out some interesting features on Mrs Black's ECG, we took the opportunity to expand our ECG skill set.
 |
| True fact: He's an ER fan! |
Analyzing the initial ECGs
There's a good bit of cardiology literature devoted to making this diagnosis (AVRT vs AVNRT) using just the surface ECG.
(If you want to do your own "deep dive" in this area, start of with these three articles, for example: Combined evaluation of bedside clinical variables and the electrocardiogram..., Electrocardiographic differential diagnosis of narrow QRS complex tachycardia, and EGC diagnosis of paroxysmal supraventricular tachycardias...,.)
If, however, you don't feel like tackling the primary literature, you can instead check out this table, summarizing the elements that distinguish AVNRT form AVRT on the ECG:
 |
| Source: Gonzalez-Torrecilla 2011 |
First let's look at Mrs Black's ECG. Are there indications of atrial activity? The best leads for this are usually II and V1. Typically the P-wave is upright in II, and inverted in V1, but a close look at those leads...
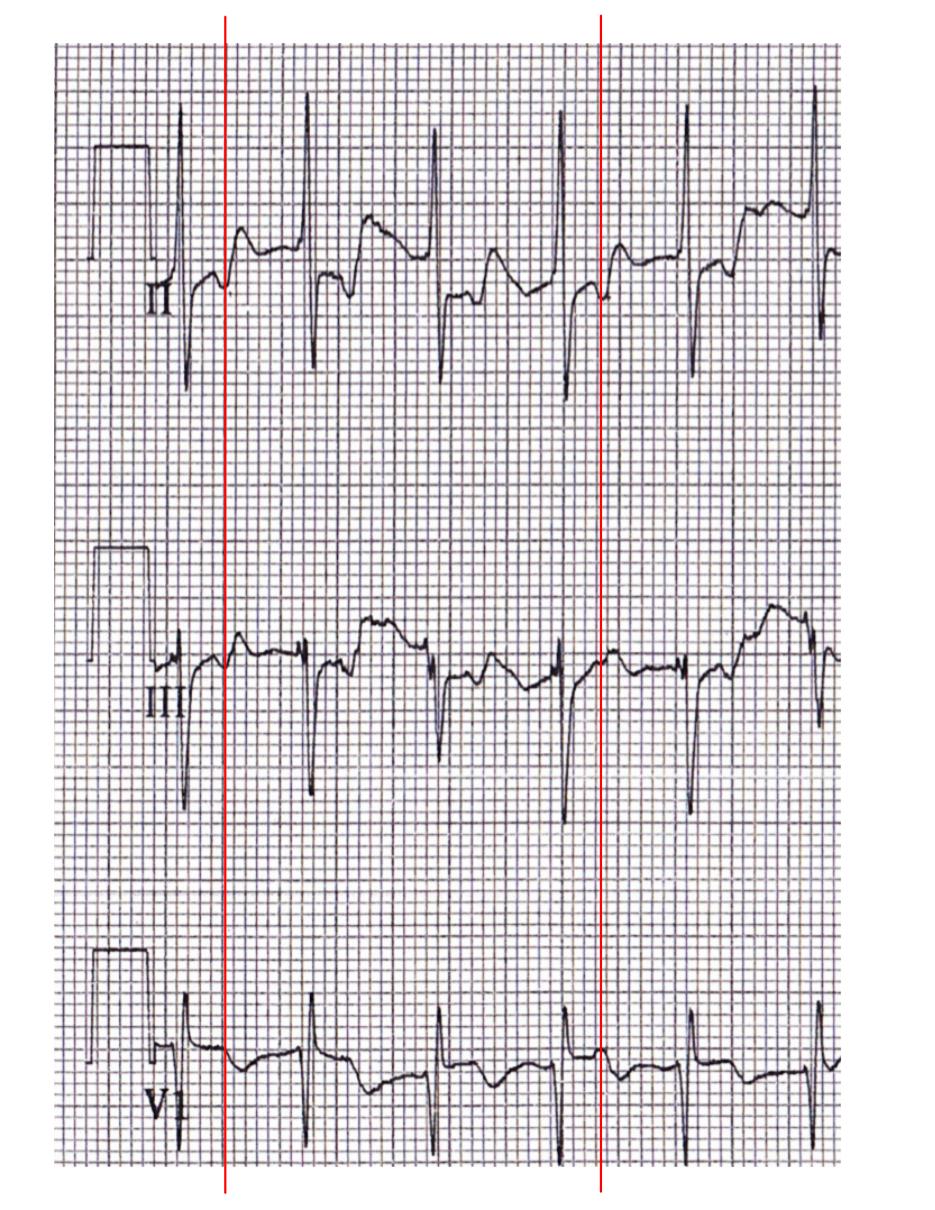 |
| Detail of "Mrs Black - before" |
These elements all suggest that this is an orthodromic AVRT, using a concealed pathway. Looks like the electrophysiologist has to go looking for a bypass tract!
Ok, what about Mr White?
 |
| You remember Mr White? He has a cool-sounding name. |
 |
| Detail of "Mr White - Before" |
 |
| Pseudo S' in AVNRT, before & after conversion. Source |
So what?
So what's the big deal? Diagnosing an AVRT versus AVNRT in the ED isn't really crucial to the immediate management. So why should you try and delve deeper into the ECG?
Well, first off, in EM we have the tough job of trying to sound smart to a lot of specialists (without ending up sounding too clever by half!). In any one shift, you'll find yourself describing an "overlapping distal radial fracture with intraarticular extension" to an orthopedist, a "perilimbal flush with consensual photophobia" to an ophthalmologist, and "He says he wants detox. Again." to the social worker. Similarly, in cardiology, we should using the language of their field, describing inverted Ps, pseudo S' waves, and such.
Furthermore, we can help point the patient in the right direction sooner. Evidence of AVRT, for example, suggests that we should talk with an electrophysiologist before discharge, arranging follow-up.
Lastly, you can't know too much about ECGs. Emergency physicians have to be experts in acute cardiology, give the pivotal role we have in the system. We can't afford to be second-best to anyone in the hospital, especially at 0300!
Thanks again to Dr Gordon for inspiring this post, and assisting with the cardiology perspective!






































